(通讯员 余紫钧)近日,我院万亮副教授与武汉大学、浙江师范大学以及日本大阪大学等国内外多所高校的研究人员共同合作,在加速分子动力学模拟新方法开发方面取得重要进展,提出了一种名为“Shuffling Accelerated Molecular Dynamics(SAMD,原子重排加速分子动力学)”的分子动力学加速方法,有望显著提升对材料微观结构演化过程的模拟能力。研究成果以“An empirical formulation of accelerated molecular dynamics for simulating and predicting microstructure evolution in materials”为题,发表于国际物理学期刊《Computer Physics Communications》。万亮副教授为第一作者兼共同通讯作者,长江大学为第一单位。

长期以来,传统分子动力学(MD)计算机模拟方法在材料科学与工程研究中被广泛应用,但其在时间尺度上的局限性阻碍了对许多重要材料演化过程(如相变、位错运动、晶界扩散等)的研究。为解决这一瓶颈,研究人员基于扩展系统动力学框架,发展出一种全新的、通用的模拟加速方法SAMD。
研究团队提出了一种名为“最近邻偏心绝对位移(NNOAD)”的物理量,用于量化原子与其最近邻原子团的几何中心的偏离程度。研究认为,所有原子的NNOAD集合可以作为一个广义反应坐标,广泛适用于描述材料中各种微观结构转变过程。该方法不再依赖于针对特定微观结构转变事件预设的反应坐标,大大增强了通用性和适用性。在SAMD方法中,每个原子的NNOAD的三个分量被分别耦合到一个额外的动力学变量上,形成一套六维空间上的扩展系统的动力学方程。系统中原子在真实三维空间的运动采用Nosé-Hoover动力学进行温度控制,而在扩展空间中,额外动力学变量遵循Langevin方程演化。这种独特的六维空间动力学组合确保了在加速微观结构转变的同时,系统仍能保持与常规MD模拟一致的动力学行为。
通过精心选择的基准问题模拟(如α-Fe晶体中碳间隙原子和单空位的扩散),研究团队确定了SAMD方法中关键参数(如耦合系数κ、额外动力学变量的阻尼系数γₛ和“驱动温度”Tₛ)的合理取值区间。研究发现,在选定的参数范围内,SAMD方法能够对不同能量势垒的结构转变(如空位迁移和间隙原子扩散)产生近乎一致的加速效果,避免了传统加速方法中因势垒高度差异导致的加速不均问题。此外,研究人员通过四个典型的材料科学问题,全面验证了SAMD方法的有效性和准确性:(1)氢在铝晶界上的偏聚:成功模拟了低温下氢原子的长程扩散和晶界偏聚过程,加速比可达数千至上万倍;(2)铝晶界的剪切力学响应:模拟了晶界DSC位错形核与扩展引起晶界滑移-迁移耦合运动的机制,在保持结构转变本质不变的前提下,显著降低了临界剪切应力,有效延长了模拟的时间尺度,加速效果估计可达约8个数量级;(3)α-Fe双晶的拉伸力学行为:再现了拉伸过程中位错从晶界形核、与晶界相互作用等一系列复杂结构转变,获得的应力-应变曲线与结构演化过程符合物理预期;(4)银薄膜表面扩散:有效模拟了表面点缺陷(空位、吸附原子等)的迁移与转化,展示了方法在研究表面动力学方面的潜力。
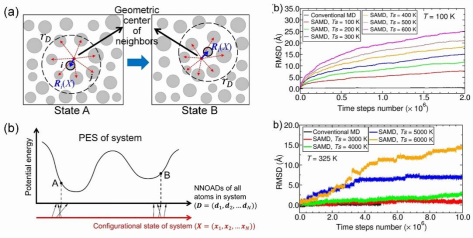
该研究提出的SAMD方法有望推动对材料变形、合金时效、辐照损伤演化、晶体气相生长等长时间尺度过程的高效原子尺度模拟研究,为深入理解材料性能演变的微观机理、设计新型高性能材料提供强大的计算支持。研究团队也指出,其理论基础的进一步完善和严格证明是未来工作的重点。通过对SAMD动力学方程的深入分析,有望更精确地确定方法参数并量化其加速能力,从而推动这一经验性方法走向更加成熟和普适的理论框架。
该研究得到了国家重点研发计划、国家自然科学基金等项目的资助。
论文链接:https://www.sciencedirect.com/science/article/pii/S0010465525004448
(审核:水涛,编辑:梁军)

